Content:
1. Introduction to the topic of enzymes
2. Mechanism of action and factors affecting enzyme activity
3. Nomenclature of enzymes
4. Enzyme inhibition
5. Use of enzymes in diagnostics
6. Overview of cofactors
_
Introduction to the topic of enzymes
Terms and structure
There are many different kinds of reactions in biological systems. Attempts to reproduce them outside the living systems (in vitro) revealed, that their speed is significantly lower. In vivo reactions are hundred to million times faster than the same reactions taking place in vitro. A major cause of this difference is the existence of specific catalysts – enzymes. Enzymes allow the existence of reactions that would not otherwise occur under conditions (temperature, pH, …) present in human body.
A catalyst is a substance, which increases the rate of a chemical reaction, but do not alter the chemical equilibrium (only shortens the time until it is achieved). Molecules of enzymes leave reaction unchanged and are not consumed.
Enzyme is a specific organic molecule speeding the reaction in biological systems (acting as biocatalyst). It enables reactions to happen under relatively low temperatures, neutral pH and atmospheric pressure, conditions that are common in our bodies. The vast majority of enzymes are proteins (exceptions are some kinds of RNA molecules called ribozymes). Apart from protein component, enzymes can also contain non-protein part. According to its presence, enzymes can be divided into:
1) Simple: containing protein part only (e.g. hydrolases like pepsin, trypsin or ribonuclease)
2) Complex: apart from a protein part called apoenzyme (that is ineffective on its own), they contain a non-protein part as well – called cofactor. Cofactor, together with apoenzyme, forms a biologically active molecule of enzyme – the holoenzyme.
Cofactor may be:
1) Metal ion: Zn2+ (e.g. alcohol dehydrogenase), Mn2+ (e.g. arginase), Fe2+, Cu2+, Mg2+
2) Organic molecule: very often a derivative of vitamin; according to the nature of its bond with apoenzyme, we further recognise:
a) Coenzymes: a non-protein organic molecule that binds to the molecule of apoenzyme freely, and thus can detach from it (e.g. NAD+, NADP+)
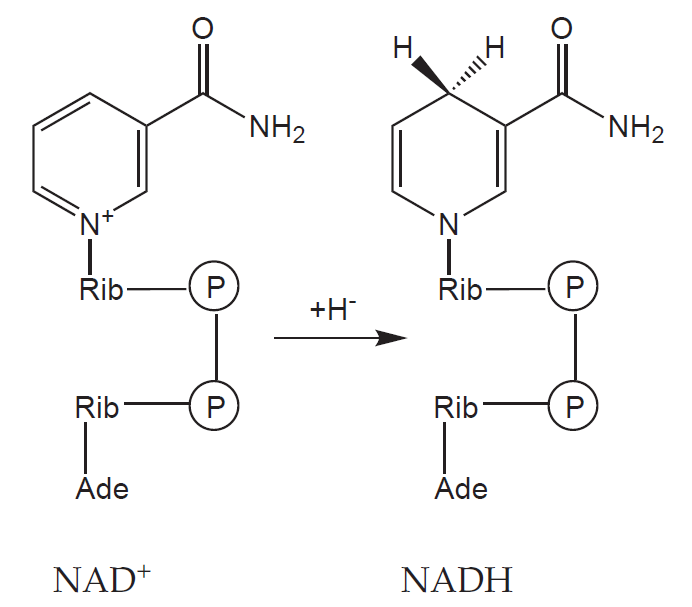
b) Prosthetic group: a non-protein organic molecule that binds to the molecule of apoenzyme tightly (e.g. heme, FAD)
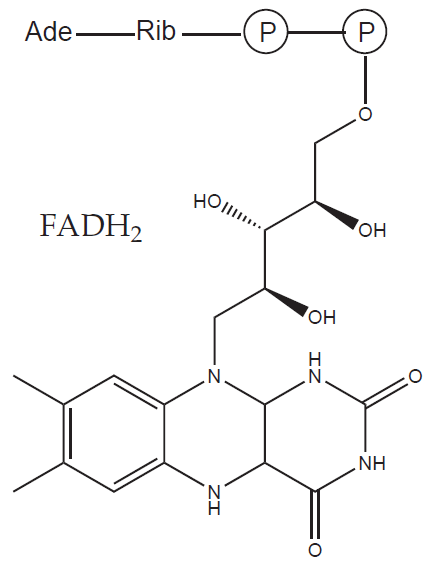
A detailed overview of cofactors and enzymes is listed at the end of this chapter.
Enzymes are made up of different number of peptide chains. Even in the case of only one chain, it can contain multiple parts called domains (of same or different enzyme specificity). Enzymes consisting of multiple chains (and thus having quaternary structure) are termed multienzyme complexes. Individual subunits usually have different specificities and are bound non-covalently. An example of multienzyme complex is the fatty acid synthase, an enzyme catalyzing the synthesis of higher fatty acids in cells.
Some enzymes (for example a lot of gastrointestinal enzymes) are produced and stored in their inactive forms termed zymogenes (or proenzymes). The reason is an effort to protect the secretory cells from the autodigestion caused by active forms of the enzymes. Zymogenes are only activated at the place, where their action is desired. Enzymes can be activated, for example, through a process of partial proteolysis, by splitting off a defined portion of the molecule. Let’s look at two examples of this process. The chief cells of the gastric mucosa secrete proenzyme pepsinogene. HCl present in gastric juice helps with the autoactivation of pepsinogen to its active form pepsin. The reaction also takes place autocatalytically, by cleaving the molecules of pepsinogen with already produced pepsin molecules. Similarly to pepsin, the production of trypsin in pancreas starts with its proenzyme trypsinogen. Enteropeptidase in the small intestine (synthesized by the cells of intestinal mucosa) split off a hexapeptide and thus create an active molecule of trypsin.
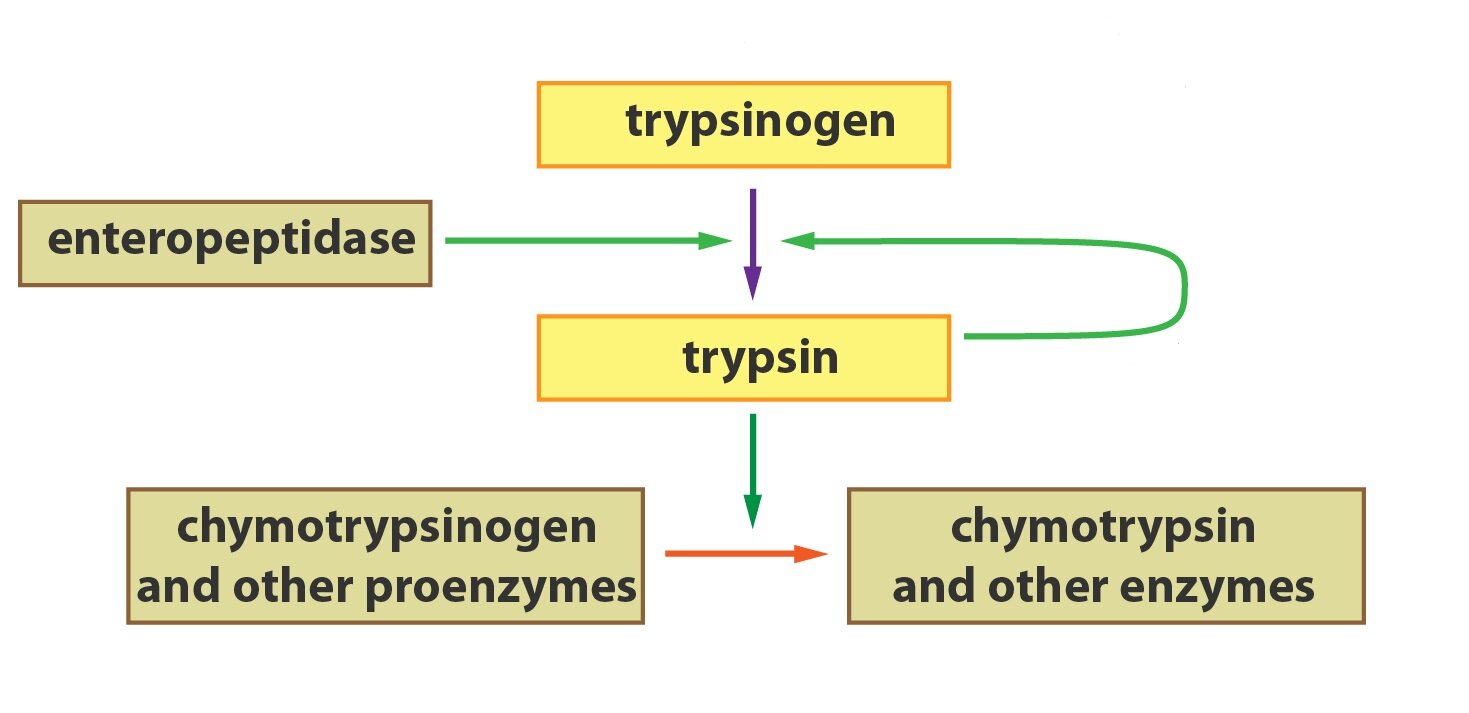
Isoenzymes and isoforms of enzymes
In the organism, there exist enzymes (called isoenzymes) catalysing the same reactions, but differing from each other in their physico-chemical properties (different substrate affinity, KM or sensitivity to inhibitors) and presence in tissues. These differences originate in genetics (isoenzymes have different primary DNA structure) and allow, for example, for a certain regulation of the reactions to be based on the different conditions in different tissues.
Very good examples are the isoenzymes catalysing the conversion of glucose to glucose-6-phosphate (a phosphorylation of glucose): glucokinase (located in hepatocytes and pancreatic β-cells) and hexokinase (located in other cells of the body). Glucokinase shows lower affinity for its substrate – glucose (expressed by KM – see further; KM of glucokinase is approximately 10 mmol/l), which means that the enzyme-catalysed reaction takes place only when the level of blood glucose is high enough (mainly after a meal). In between meals, when the blood glucose is normal, the glucokinase is only slightly active and the liver leaves glucose for other tissues (containing hexokinase with KM value around 0.1 mmol/l). For details see Subchapter 2/9.
Apart from isoenzymes, body produces so-called isoforms of enzymes as well. They are multiple forms of enzymes derived from the same gene (so the primary DNA structure is the same), but subjected to different posttranslational modifications or alternative splicing. Resulting from this, isoforms of enzymes can catalyze different reactions.
_
Mechanism of action and factors affecting the enzyme activity
Enzymes, similar to other catalysts, work by reducing the value of activation energy (see Subchapter 2/4).
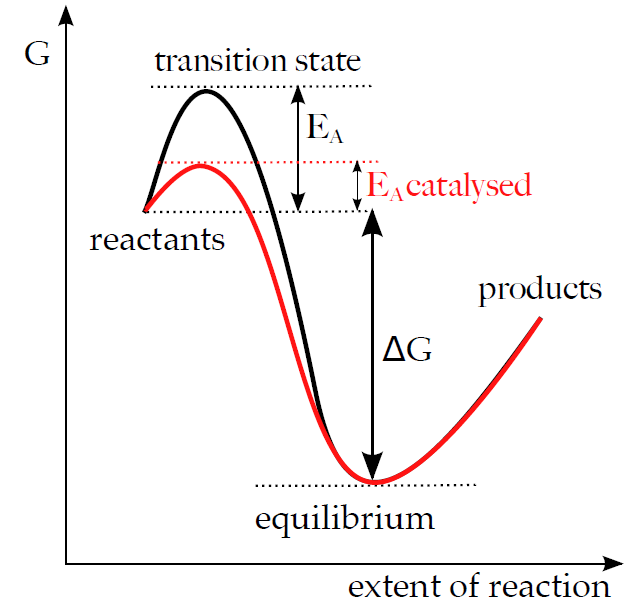
An enzyme-substrate complex (E-S) forms in the first step of the catalysis. This reaction is usually very fast and reversible. Subsequently, the enzyme catalyses the conversion of a substrate to its product and the complex E-S changes to E-P complex (enzyme-product). E-P complex breaks down and releases the product in a slow and usually irreversible manner. Thus the reaction is divided into several successive steps, which contain one or several E-S transition states. The activation energies necessary for the process of their synthesis and the conversion of E-S to E-P are lower than the EA involved in the direct conversion of substrate to product, although the overall ΔG of both reactions stays the same.
Substrate – enzyme interactions
Substrate interacts with the molecule of enzyme in an area called the active site (center). It consists of:
1) Binding site of the enzyme
The binding site of the enzyme is small, spatially delimited part of an molecule containing accurately arranged functional groups (-SH, -OH, acidic and basic amino acids) whose position corresponds with the structure of the substrate. The interactions between enzyme and substrate are mostly non-covalent (H- bridges, electrostatic and hydrophobic interactions, van der Waals forces), covalent bonds are rare.
2) Catalytic site
The catalytic site contains other groups, which are responsible for the catalytic activity of enzyme. These groups often come from the molecule of cofactor.
It is usually difficult to precisely differentiate between binding and catalytic site of the enzyme.
Apart from the active site, there can be other sites on the surface of the enzyme called allosteric, which enable the regulation of enzyme through different effectors (inhibitors or activators).
The original model of substrate – enzyme interaction (later named the lock and key theory), created by a German Nobel laureate Emil Fischer at the end of 19th century assumed, that the molecule of substrate fits precisely into the molecule of enzyme. This model was revised in 20th century by American biochemist Daniel Koshland. He assumed that the molecule of substrate is able to induce a conformational change of the binding site (and vice versa). The exact shape of “lock and key” is achieved only after the binding. It appears that this induced fit theory suits the description of the interaction between enzyme and substrate better than the original model.
Enzyme specificity
Enzyme specificity implies different limitations on the range of action of a particular enzyme. We distinguish:
1) Substrate specificity
A substrate specificity means, that an enzyme acts only on a limited range of substrates and cannot catalyze reactions involving other substrate. There are various extents of substrate specificity:
a) Absolute
Enzyme catalyses the conversion of only one particular substrate, but do not catalyze the conversion of other substrates, even when these are derivatives of the original substrate. An example is urease, an enzyme catalyzing the reaction: urea + H2O → CO2 + 2 NH3. Urease however, will not catalyze the hydrolysis of methylurea or thiourea.
b) Group specificity
Represents a more frequent form of substrate specificity, where the enzyme catalyses reactions involving several similar substrates (usually having the same functional groups). The affinity for each substrate can vary (KM for each substrate can be different). As an example we can take carboxypeptidase B, an enzyme hydrolyzing peptides from their carboxy – terminus and preferentially cleaving peptide bonds with charged amino acids (arginine, lysine).
2) Linkage specificity
Linkage specificity means that the enzyme will act on a particular type of chemical bond regardless of the rest of the molecular structure. Lipases, for example, are enzymes hydrolyzing different kinds of lipid molecules.
Apart from these types of specificity, a lot of enzymes are stereospecific as well, meaning that they only attack certain conformational isomers of substrates (e.g. only L- or only D- form). It is probably due to the requirement to bind to the substrate at, at least three specific places of the active site of the enzyme (that is a chiral compound) – the other stereoisomere would not bind.
Factors affecting the enzyme activity, kinetics of one-substrate reactions
Substrate concentration
Enzyme kinetics of one-substrate reactions had been studied by Leonor Michaelis and Maud Leonora Menten. Laws of general kinetics, discussed in Chapter 2: Subchapter 4. (concerning equilibrium constant, reaction rate, Gibbs energy), apply here as well.
In the case of catalyzed one-substrate reactions we assume that they take place in two steps:
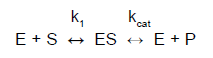
where kcat is the reaction rate of the catalyzed reaction. It is also called a turnover number and it indicates the number of molecules of substrate converted by one molecule of enzyme per unit of time (typically one second). The values range from 4.107 (for catalase) till 0,5 (for lysozyme).
The maximal reaction rate of the catalyzed reaction can be expressed as:
vmax = kcat [E]t
where [E]t is the total enzyme concentration.
Michaelis-Menten equation describes the dependence of (initial) velocity v0 on the reaction rate kcat and on the E-S equilibrium as well:
v0 = Δ[P] / Δt =
= (kcat [E]t [S]) / (KM + [S])
We assume that the amount of enzyme is constant (and its concentration is significantly lower than the concentration of substrate) and we also assume a presence of a certain pseudo-steady state, during which the E-S concentration changes much more slowly than the concentrations of S and P.
Substituting the equation (2) to the equation (3) we get:
v0 = (vmax [S]) / (KM + [S])
KM (Michaelis constant) is experimentally defined, as the concentration of substrate at which the reaction rate of the enzymatically catalyzed reaction is equal to the half of the maximum rate. KM thus expresses the affinity of substrate and enzyme. The lower the value of KM, the higher the affinity (a lower [S] is necessary to saturate half of the enzyme molecules).
v = vmax / 2
The graph above shows that the dependence of the rate of the enzyme-catalysed reaction on the substrate concentration has a hyperbolic shape. At low substrate concentrations, the curve corresponds to the 1st order kinetics (linear increase). This, almost linear increase, cannot continue indefinitely, due to the limited amount of binding sites on the molecules of enzymes. At sufficiently high substrate concentration, after saturating all available binding sites of all enzyme molecules, a maximal speed (vmax) is reached and remains approximately constant. This part of the curve corresponds to the 0thorder kinetics (the speed is no longer dependent on the substrate concentration).
The value of KM can be determined using graphical methods directly from the hyperbolic curve, but it is not often used. The use of reverse values 1/v and 1/[S] (so-called double reciprocal plot according to Lineweaver and Burk) is more accurate. From equation (4):
1/v = (KM + [S]) / (vmax [S])
We can rewrite the equation in the form of a line equation y = a* x + B
1/v =(KM / vmax). 1 / [S] + 1 / vmax
Temperature
By applying high temperature, enzymes, due to their protein nature, denature (undergo a conformational change that causes the destruction of all, but the primary structure and thus impair the active site as well). Denaturation usually occurs at temperatures around 55-60 °C and causes a drop in or a complete halt of the reaction. Low temperatures, on the other hand, can slow down reactions as well, by reducing the activity of enzymes.
There exist a temperature, called the temperature optimum, at which the enzyme has the maximal activity (in the case of majority of the human enzymes it lies around 37 °C).
pH (H+ concentration)
Similarly to the temperature changes, enzymes are also sensitive to the changes in pH. Extreme values (both low and high) can lead to the denaturation of the enzyme molecules. In addition, the concentration of H+ affects the ionization of acidic and basic groups of the enzyme and substrate. Each enzyme has its pH optimum, which is a value of pH when the enzyme activity is the highest.
Activators and inhibitors
Other factors affecting the activity of enzymes include activator and inhibitors. Due to their great importance, we discuss them in a separate section of this chapter.
Expressing the enzyme activity
In practice, instead of determining the amount of substance or concentration or the enzyme, we measure its catalytic activity. It shows the rate of the reaction under given conditions.
Originally, the international unit of enzyme activity (U) had been used. 1 U stands for the amount of enzyme that catalyzes, under standard conditions, the conversion of 1 μmol of substrate in 1 min.
Newer unit is katal (kat). 1 katal is the amount of enzyme that, under standard conditions, catalyzes the conversion of 1 mol of substrate in 1 second. However, the unit is too large for practical use. In medicine we mostly use its fractions – microcatal (μkat) – 10-6 kat and nanokatal (nkat) – 10-9 kat.
To convert the international unit of enzyme activity to katal, we use the following equation: 1 kat / 6x 107 U.
_
Nomenclature
Trivial nomenclature
The formerly used term ferments that was based on the fact that enzymes are involved in the process of fermentation, is no longer used. First discovered enzymes were given names according to their source or method of discovery. Their names are therefore without any relation to the reaction mechanism. Many end with –in ending.
An example is pepsin, discovered in gastric juices (gr. pepsis – digestion) or ptyalin, found in saliva (gr. ptyalon – saliva).
Recommended nomenclature
Simpler than systematic nomenclature, but yet being systematic in a way, the recommended nomenclature is often used in routine practice. The name is made by combination of:
1) Substrate + ase: e.g. amylase (catalysing the hydrolysis of amylose)
2) Type of the reaction + ase: e.g. dehydrogenase
Systematic nomenclature
Systematic nomenclature has been introduced by IUB (International Union of Biochemistry). Every enzyme has its own EC (Enzyme Commission) number, which consists of four digits separated by commas – x.x.x.x. The first digit denotes one of the six main enzyme classes, second and third stand for a subgroup and a subsubgroup and the last indicates the precise position of enzyme within the subgroup (and thus completely characterises the enzyme). We distinguish the following six major classes of enzymes:
1) Oxidoreductases
2) Transferases
3) Hydrolases
4) Lyases (synthases)
5) Isomerases
6) Ligases (synthetases)
1) Oxidoreductase
Oxidoreductases catalyse reactions involving the oxidation of one and the reduction of the other component. They often use cofactors – e.g. NAD+, NADP+, FAD or heme.
Examples are:
a) Oxidases, peroxidases
b) Oxygenases: they bring oxygen molecule into the molecule, either as a -OH group (monooxygenases, also called hydroxylases) or as a O2 (dioxygenases)
c) Dehydrogenases: they oxidise the substrate by eliminating H-atoms; they are often abbreviated as DH (e.g. lactate dehydrogenase – LDH, alcohol dehydrogenase – ADH)
d) Desaturases
2) Transferases
Transferases participate on the transfer of various groups (amino-, acyl-, methyl-, glycosyl-, phosphoryl-,…) from one molecule to another.
Examples are:
a) Transaminases (aminotransferases): participate in the transfer of -NH2 group
b) Kinases (phosphotransferase): participate in the transfer of phosphate group from ATP (or other nucleoside triphosphates)
c) Transaldolases, transketolases
3) Hydrolases
Catalyze the hydrolytic reactions (the cleavage of bonds in molecules with the help of water molecule).
Examples are:
a) Lipases, phospholipase
b) Disaccharidases (saccharase, maltase, lactase)
c) Proteases, peptidases (pepsin, trypsin)
d) Esterases
e) Phosphatases
4) Lyases (synthases)
Catalyze the removal of a certain group from the substrate (elimination reaction) by means other than hydrolysis (non-hydrolytic cleavage, for example of bonds between C-C or C-N). They also catalyze additions to the double bond and synthetic reactions without ATP consumptions. Examples are:
a) Decarboxylases
b) Aldolases
c) Dehydratases, hydratases
5) Isomerases
Catalyse changes within one molecule of substrate (intramolecular changes), so that the product is an isomer of the substrate.
Examples are:
a) Epimerases: change the position of one –OH group in the molecule
b) Mutases: change the position of a phosphate group within the molecule
6) Ligases (synthetases)
Catalyse synthetic reactions associated with the ATP hydrolysis (coupling of exergonic and endergonic reactions).
Examples are:
a) Carboxylases
b) DNA-ligases
_
Enzyme inhibition, importance in pharmacology
There are many substances able to affect enzyme function in terms of increase (activators) or decrease (inhibitor) in its activity. The inhibition of enzyme activity is one of the most important regulatory mechanisms in living systems. Apart from these naturally occurring phenomena, the inhibition of specific metabolic pathways underlies the effect of many drugs. It is therefore important to know the mechanism of their inhibition.
Based on the reversibility of the effect, there are two main types of inhibition:
1) Reversible
Reversible inhibition can be suppressed (or reversed). Inhibitor binds non-covalently (through weak chemical interactions) either to the enzyme’s active site or outside of it. The effect of inhibitor can be eliminated for example through increased supply (concentration) of substrate or through dialysis.
a) Competitive
Competitive inhibitor competes with the molecule of substrate for the active site of the enzyme. The inhibitor is therefore often structurally similar to the molecule of substrate; it binds to the enzyme, but its unable to undergo enzyme-catalyzed reactions. Increasing the substrate concentration displaces the inhibitor from the active site and thus suppresses its effect. Competitive inhibitor does not affect vmax, it only delays its attainment (until the inhibitor is displaced by an increased concentration of substrate. KM increases (seemingly, the affinity of an enzyme to substrate is lowered).
b) Non-competitive
Non-competitive inhibitor binds outside the binding site for substrate (the site of its binding is sometimes referred to as an allosteric site). Through its binding, it changes the conformation of the enzyme to such extent that it affects the conformation of the active site as well, preventing the binding of the substrate. Increasing the substrate concentration does not suppress the inhibition (as the substrate does not bind to the allosteric site, there exist no competition for the binding site). The only way to eliminate the inhibition is to remove the inhibitor (e.g. through dialysis).
Because none of the enzyme-inhibitor complexes (or enzyme-inhibitor-substrate complexes) is catalytically active, the amount of enzyme (that would be available to substrate) decreases. That causes a decrease in vmax.KM on the other hand, is not affected.

c) Acompetitive
In this type of inhibition, the inhibitor binds only to an enzyme-substrate complex creating a ternary complex of enzyme-inhibitor-substrate.
The result is a decrease in both vmax (because the complexes are not enzymatically active) and KM, but their ratio stays the same. Inhibitor has little effectiveness at low substrate concentrations, because there are not enough E-S complexes to which the inhibitor binds.
2) Irreversible
Irreversible inhibition involves a covalent modification of the enzyme molecule. Inhibitor covalently binds, either to the active site of the enzyme or outside of it, and thus it is not possible to suppress the inhibition (e.g. by dialysis or by increasing the substrate concentration).
Examples are heavy metals (Ag+, Hg2+,…) or organophosphates (or their derivative such as nerve gases sarin or tabun).
Another phenomenon is an excess-substrate inhibition. When the concentration of substrate is too high, the individual molecules compete with each other for the active sites. It results in a slight decrease in vmax at high [S].
Allosteric regulation of enzyme activity
Many of the “rate-limiting” (or regulatory) enzymes of the metabolism are allosteric. Allosteric regulation of their activity is one of the most important ways to regulate metabolic pathways.
The surface of an allosteric enzyme contains, apart from an active site, other site called allosteric (gr. allos – other) through which it may be affected by modulators (activators or inhibitors). When an allosteric modulator binds to this site it causes a conformational change of the enzyme molecule leading to the change in affinity for substrate or other ligands. Most of the allosteric enzymes are oligomers (they are composed of more subunits). The binding of the modulator to one subunit affects (through conformational change) other subunits as well. We recognize two types of allosteric regulation:
1) Homotropic: modulator functions at the same time as a substrate. A well-known example (though not being an enzyme) is a molecule of O2, which is a homotropic allosteric modulator of hemoglobin.
2) Heterotrophic: modulator and substrate are different molecules. To build on the previous example, CO2 would be a heterotropic allosteric modulator of hemoglobin.
Allosteric enzymes show sigmoidal kinetics
As an example, we take a reaction affected by a homotropic allosteric activator. At low concentrations of substrate, the reaction proceeds very slowly, because only a handful of enzyme molecules are occupied by substrate. The turnover occurs, when the enzymes start to have at least one subunit bound to the molecule of substrate (acting as an activator). This also increases the affinity of other subunits to substrate molecules. As seen in figure above, the reaction rate rises sharply at this substrate concentration. The more subunits the enzyme molecule consists of, the sharper is the onset of the effect of increased substrate concentration. Enzyme works according to “all-or-nothing” principle. The reaction almost stops under certain [S], but above it, it quickly achieves vmax (where all binding sites are saturated). This characteristic of the allosteric enzymes is very advantageous, since it allows to quickly “turn on” or “off” the reaction and thus the whole metabolic pathway.
_
The use of enzymes in diagnostics
Measuring the activity of various enzymes in body fluids is often used in diagnostics to determine the place and extent of tissue damage. Apart from diagnostic contribution, the enzyme activity can give us prognostic information as well (mostly evaluated from the changes in the level of enzymes in time).
We usually measure the enzyme activity directly in the blood plasma:
1) Enzymes specific of the plasma – e.g. coagulation factors
2) Secretory enzymes – that get into the blood in various amounts (e.g. pancreatic amylase or lipase).
3) Intracellular enzymes carrying out different functions within cells. They usually enter the blood after damage to the tissue.
When evaluating the levels of enzyme activity, it is important to know their:
1) Intracellular localisation
Some enzymes are found only in certain cellular compartments and can therefore act as their markers. For example an enzyme cytochrome oxidase is specific for mitochondria. According to the spectrum of enzymes found in the blood, we can deduce how severe is the damage done to the tissues. If only cytosolic enzymes enter the blood, the impairment is milder than in the case of occurrence of mitochondrial enzymes.
2) Organ and tissue distribution
Similar to the distribution of the enzymes in cellular compartments, enzymes differ in their distribution among tissues and organs. Some enzymes are more or less organ specific. These are typically various kinds of isoenzymes or isoforms of enzymes. An example might be isoenzymes of creatine kinase – CK-MB that is typical for myocardium or CK-MM found in skeletal muscle.
The most commonly measured enzymes
1) Liver
a) Markers of hepatocytes lesion:
1. ALT (alanine aminotransferase) – located mostly in cytoplasm
2. AST (aspartate aminotransferase) – located mostly in mitochondria
AST, as a predominantly mitochondrial enzyme, is released only after more severe damage to the liver cells. According to AST/ALT ratio, we can estimate the extent of the liver damage.
b) Markers of cholestasis:
1. ALP (alkaline phosphatase)
2. GMT (gamma-glutamyltransferase) – increased activity is also present in chronic alcoholic liver lesions.
2) Pancreas
a) Pancreatic amylase –less specific than lipase
b) Pancreatic lipase
3) Muscle tissue
To distinguish between the possible sites of lesion we measure the activity of the creatin kinase isoenzymes:
a) CK-MM – found in skeletal muscle
b) CK-MB – found in myocardium
c) CK-BB – found in brain
Apart from assessing the CK-MB levels, in the case of myocardial infarction, we measure the levels of following enzymes:
a) Troponin I and T, which are currently the most specific markers of cardiac damage. Their concentration rises approximately 12 hours after infarct.
b) Myoglobin, which is less specific, but achieves its peak concentration in blood already 2 hours after a heart attack.
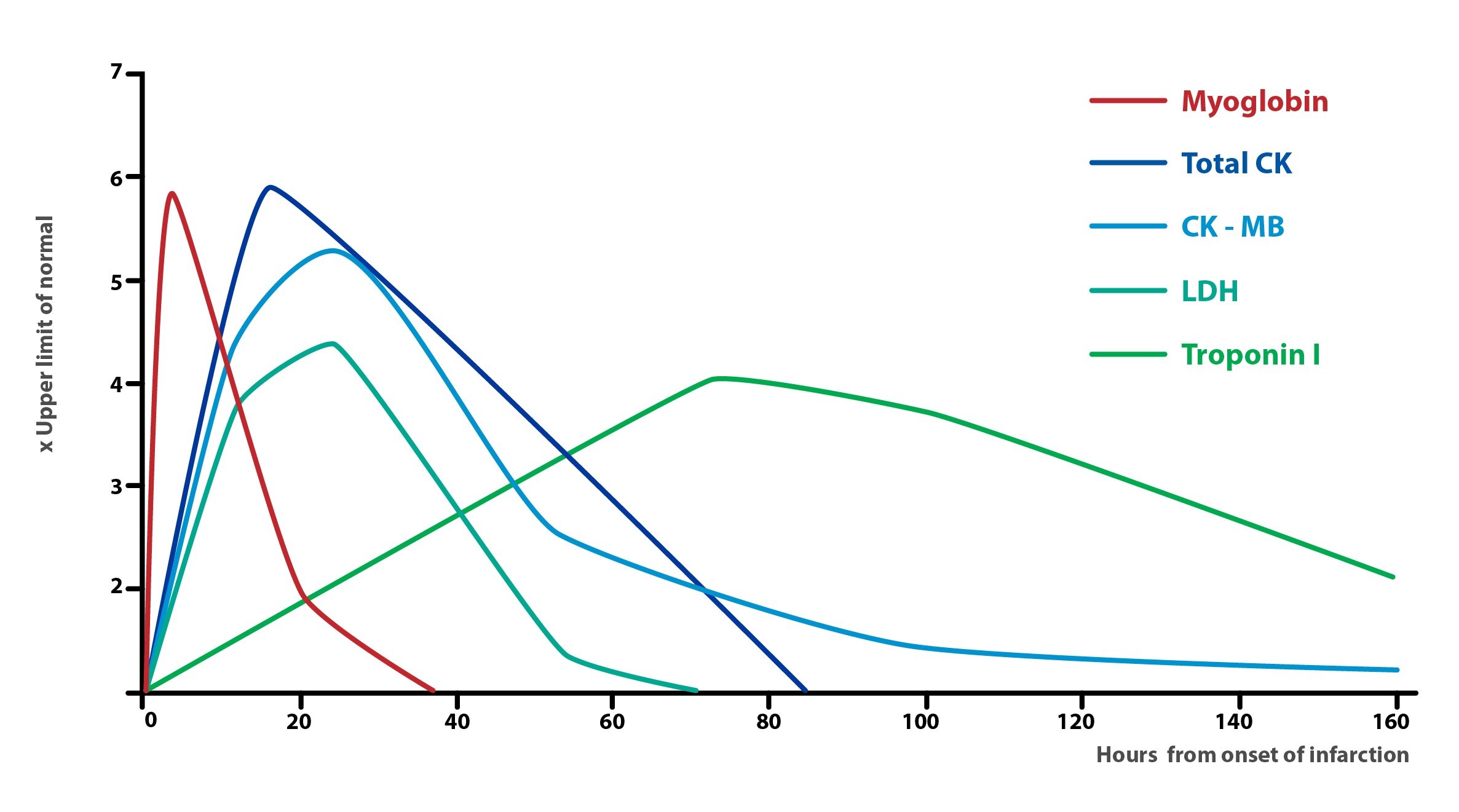
4) Bone tissue
a) ALP (alkaline phosphatase) – located in osteoblasts (so-called bone fraction), but also in liver (so-called liver fraction), GIT or kidney cells.
b) ACP (acid phosphatase) – located in osteoclasts and prostate cells
Other diagnostic enzymes include:
1) Lactate dehydrogenase isoenzymes (LD, LDH)
a) LD-1 – found in cardiomyocytes and erythrocytes
b) LD-2 – found in RES
c) LD-3 – found in lungs
d) LD-4 – found in kidneys, pancreas and placenta
e) LD-5 – found in liver and skeletal muscle
| Enzyme | Organ/tissue |
| α-amylase (AMS), pancreatic lipase (LPS) | Pancreas |
| Alkaline phosphatase (ALP) | Bone, liver, GIT, kidney, placenta |
| Acid phosphatase (ACP) | Bone, prostate |
| Creatine kinase (CK) | Skeletal muscle, myocardium |
| Lactate dehydrogenase (LD) | Myocardium, liver, skeletal muscle, kidneys, erythrocytes |
| Alanine aminotransferase (ALT) | Liver |
| Aspartate aminotransferase (AST) | Myocardium, liver, skeletal muscle |
| Gamma-glutamyltransferase | Liver |
_
Overview of enzyme cofactors
1) Metal ions and trace elements
|
Cofactor |
Examples |
| Zn2+ | Peptidases, alcohol dehydrogenase |
| Mg2+ | ATP dependent enzymes, phosphohydrolases |
| Mn2+ | Superoxide dismutase, arginase |
| Fe2+/ Fe3+ | Cytochromes, catalase, peroxidases |
| Cu2+ | Cytochrome oxidase, amino oxidases |
| Mo2+ | Xanthine dehydrogenase |
_
2) Organic compounds as cofactors
a) Cofactors of oxidoreductases
|
Cofactor |
Precursor vitamin |
Localisation / function |
| NAD+, NADP+ | Nicotinic acid | Respiratory chain, FA synthesis |
| FAD, FMN | Riboflavin (B2) | Respiratory chain |
| Ubichinon/ubichinol | Respiratory chain | |
| Heme | Cytochromes |
_
b) Cofactors of transferases
|
Cofactor |
Precursor vitamin |
Localisation / function |
| ATP, GTP | Thiamine (B1) | Transfer of phosphate residue |
| TDP (thiamine diphosphate) | Thiamine (B1) | Transfer of carbon fragments (oxidative decarboxylations) |
| PALP (pyridoxal phosphate) | Pyridoxine (B6) | Transfer of -NH2 group (transaminations), decarboxylations of amino acids |
| THF (tetrahydrofolate) | Folate (folic acid) | Transfer of one-carbon fragments |
| CoA (coenzyme A) | Pantothenate | Transfer of acyls |
| PAPS (phosphoadenosine phosphosulfate) | Transfer of sulphates | |
| SAM (S-adenosyl methionine) | Methylations | |
| B12-complex | Cobalamin (B12) | Transfer of -CH3 group |
_
c) Cofactors of lyases
|
Cofactor |
Precursor vitamin |
Localisation / function |
| PALP (pyridoxal phosphate) | Pyridoxine (B6) | Decarboxylations |
_
d) Cofactors of ligases
|
Cofactor |
Precursor vitamin |
Localisation / function |
| ATP | ||
| Carboxybiotin | Biotin | Transfer of CO2 (carboxylations) |
_ Subchapter Author: Petra Lavríková ![]()