Content:
1. Introduction to the metabolism of proteins and amino acids
2. Amino acid degradation
3. Degradation of amino acid carbon skeletons
4. Formation of non-essential amino acids in the human body
5. Important derivatives of individual amino acids
_
Introduction to the metabolism of proteins and amino acids
Proteins are the most important and the most abundant biomolecules in the human body – a total protein amount corresponds to 14 kg (true for man weighing 70 kg). A summary of all amino acids in the body is called as amino acid pool. How is this pool formed? And how do we use it?
An adult man breaks down approximately 300-500 g of proteins to amino acids per day, this event is called as proteolysis. Approximately the same amount of amino acids is incorporated into proteins at process called proteosynthesis. Another source of amino acids, the proteins contained in the food, representing approximately 70-100 g per day. The final source of amino acids is a pool of biosynthesis of non-essential amino acids forming amino acids 30-40 g daily. Around 120 g of amino acids are degraded per day. In this process, amino acid skeleton can be divided into an amino group (-NH2 and other nitrogen atoms) and carbon chain – each of them has a completely different fate that will be described later in this section.
Amino acids may also serve as precursors to many important substances for the body – e.g. biogenic amines, heme or purines and pyrimidines.
The half-life of proteins is markedly different from protein to protein and therefore is not possible to give its average value. Generally, the structural proteins are more permanent (and therefore have a longer half-life), while the molecules of many enzymes exist very shortly – only a few tens of minutes or hours.
Proteolysis is a complete protein degradation to free amino acids. Proteases and peptidases (proteolytic enzymes) are found not only in the gastrointestinal tract , but also in each cell – lysosomes. The proteolytic enzymes are classified into:
1) Exopeptidases – amino- and carboxypeptidases – are enzymes cleaving proteins / peptides from their chain termini
2) Endopeptidases – trypsin, chymotrypsin or pepsin – are enzymes cleaving peptide bonds inside protein / peptide chains
Some proteins are degraded by ubiquitin-proteasome complex. Ubiquitin, a small protein, occurs in all eucaryotic cells. Protein designated for degradation in the proteasomes is marked by ubiquitin. This process is called as ubiquitination (optionally polyubiquitination – if multiple molecules of ubiquitin is present).
Historical correlation:
Aaron Ciechanover, Avram Hershko and Irwin Rose received the Nobel Prize for chemistry (The discovery of ubiquitin-mediated protein degradation) in 2004.
Essential and non-essential amino acid
Human body is able to synthesize only certain amino acids (non-essential, dispensable). The other amino acids (essential, indispensable) must receive in food. Here’s an overview:
Essential amino acids
1) Branched: Val, Leu, Ile
2) Aromatic: Phe, Trp
3) Basic: Lys
4) Sulphur-containing: Met
5) With hydroxyl group: Thr
Conditionally essential amino acids
Arg, His
Non-essential amino acids
Gly, Ala, Ser, Pro, Cys, Tyr, Asn, Gln, Asp, Glu
Important reactions in amino acid metabolism
1) Decarboxylation means a removal of the carboxyl group – biogenic amines are formed
2) Transamination means an exchange of amino group with 2-oxoacid – 2-oxoacids are formed
3) Oxidative deamination means an oxidative removal of amino group – 2-oxoacids are formed
4) Peptide bond formation – peptide and protein generation
Defects in amino acid metabolism
In humans, there are many different genetic defects in amino acid metabolism. Some their intermediates accumulate in the body causing defective development of the nervous system that often results in mental retardation.
_
Amino acid degradation
There are 20 (21 if we include selenocysteine) basic proteinogenic amino acids which may be inserted into protein molecules during the translation process. Catabolism of their carbon skeletons covers approximately 10-15 % of the energy requirements of the body. Amino acids may also serve as substrates (precursors) for the biosynthesis of other nutrients – carbohydrates (gluconeogenesis) and lipids.
Now we will discuss separately the fate of amino nitrogen and metabolism of amino acid carbon skeleton.
Removal of amino group
Removal of amino group is a crucial step in the amino acid catabolism. The nitrogen of the amino groups (amino nitrogen) can not be used for energy production and must be removed from our body. The first way is an amino nitrogen conversion to a urea (about 95 %), followed by urea excretion from the body via the urine. The second way is amino nitrogen releasing from glutamine in the form NH3/NH4+ in the tubular cells of the kidney (about 5 %).
Transamination
Transaminations are freely reversible reactions catalyzed by transaminases (aminotransferases). Amino group of α-amino acid is exchanged with oxo group of 2-oxoacid during transamination – from the amino acid, this produces 2-oxoacid, while from the original 2-oxoacid, amino acid is formed.
The amino group is transferred by cofactor pyridoxal phosphate (PLP, derivative of vitamin B6) to oxoacid (Schiff´s base formation).
Most of the amino acids undergo transamination in their degradation. Enzymes aspartate aminotransferase (AST) and alanine aminotransferase (ALT) are concrete examples of transaminases that are normally detected as markers of potential damage to the liver cells. Catalyzed reactions of AST and ALT are presented in the scheme.
The resulting 2-oxo acids (oxaloacetate and pyruvate) are involved in energy metabolism within cells.
But there are exceptions (e.g. threonine) that is not degraded by transamination.
Glutamate / glutamine conversion
Conversion of carboxy group in glutamate (in the side chain) to amide group in glutamine is catalyzed by cytosolic enzyme glutamine synthetase. ATP and NH4+ are also needed. This reaction is used in the cells of the CNS as the main detoxification mechanism removing toxic NH3 from brain tissue. Emerging glutamine is the most important transport form of amino nitrogen (ammonia) in the blood – provides transport from extrahepatic tissues by the blood to the liver and kidneys. Glutamine has the highest plasma concentration of the amino acids – 0.6 mmol/l (alanine – 0.3 mmol/l). Two amino groups / ammonia are “stored” in glutamine molecule. Glutamine is able to bring the ammonia to different biosynthetic processes – for example in synthesis of purine bases.
Mitochondrial enzyme glutaminase catalyzes the release of NH3 from glutamine (hydrolytic deamination, frequently occurs in hepatocytes and kidney tubule cells). The ammonia produced in the liver mitochondria enters the urea cycle. In the kidneys, ammonia is excreted in the urine, where it serves as a buffer.
Oxidative deamination
During oxidative deamination amino group is converted to keto group with simultaneous release of NH3. Glutamate is the only one amino acid that is deaminated with sufficient speed in the human body. Glutamate dehydrogenase catalyzing the oxidative deamination of glutamate is found in the mitochondrial matrix (mainly liver cells). This process demonstrates the following reaction:
Glutamate + NAD+ → α-ketoglutarate + NH4+ + NADH + H+
The resulting NH4+ enters the urea cycle and α-ketoglutarate may be used in the transamination or Krebs cycle. The mentioned reaction is fully reversible – glutamate can be synthesized from α-KG and NH4+ .
We can conclude that most of the amino acid undergoes transamination in its degradation and that the majority of amino nitrogen from amino acids is directly or indirectly concentrated in the molecule of glutamate / glutamine. Amino nitrogen is subsequently released in glutaminase and glutamate dehydrogenase reaction.
Urea (ornithine) cycle
Ammonia toxicity
Ammonia is a polar substance freely passing through physical barriers, as well as the blood-brain barrier. When its concentration increases in the body, balance of many important reactions is altered. Consider the following examples:
Glutamate + NAD+ → α-ketoglutarate + NH4+
Glutamate + NH4+ + ATP → glutamine + ADP + Pi
When an excess of ammonia, the glutamine concentration is gradually increasing. But glutamine formation also consumes α-ketoglutarate of the Krebs cycle – speed of this pathway is gradually decreasing and thus the production of energy in cells. The plasma ammonia concentration should not exceed 35 μmol / L. In the human body, most of the toxic ammonia is converted to urea by reactions of urea cycle.
Reactions in urea cycle
Urea, a non-toxic compound, is transported via the bloodstream to the kidneys where it is excreted with the urine. Urea cycle is located in the matrix of mitochondria and cytosol of liver cells. This pathway is an energy-consuming process in which the three substrates enter – ammonia, carbon dioxide (bicarbonate) and aspartate (its amino group). Mitochondrial carbamoyl phosphate synthetase I is the regulatory enzyme. Ornithine cycle communicates with the Krebs cycle via oxaloacetate and fumarate.
Urea formation involves five reactions:
1) Carbamoyl phosphate formation is catalyzed by mitochondrial carbamoyl phosphate synthetase I:
NH4+ + HCO3– + ATP → carbamoyl phosphate + 2 ADP + Pi
2) Citrulline formation is catalyzed by ornithine transcarbamoylase:
Ornithine + carbamoyl phosphate → citrulline + Pi
Citrulline is passed into the cytosol.
3) Argininosuccinate formation is catalyzed by argininosuccinate synthetase:
Citrulline + Asp + ATP → argininosuccinate + AMP + PPi
4) Argininosuccinate break down is catalyzed by argininosuccinate lyase:
Argininosuccinate → arginine + fumarate
5) Hydrolysis of arginine is catalyzed by arginase:
Arginine + H2O → ornithine + urea
Ornithine returns into the mitochondrial matrix.
The urea cycle is closely linked to the Krebs cycle – from emerging fumarate becomes aspartate. How does this relationship work? The fumarate is first hydrated to malate which is converted to oxaloacetate by oxidation. Enzyme aspartate aminotransferase catalyzes transamination between glutamate and oxaloacetate, resulting aspartate enters ornithine cycle. Glutamate is produced by transamination of degraded amino acids which transmit their amino groups on the α-ketoglutarate molecule.
Regulation of ornithine cycle
Carbamoyl phosphate synthetase I, the main regulatory enzyme of ornithine cycle, is activated by N-acetylglutamate. Enzyme N-acetylglutamate synthetase catalyzes the reaction between AcCoA and glutamate which produces N-acetylglutamate. The amino acid arginine increases the enzyme activity. Transcription of urea cycle enzymes is increased in high-protein diet or by increasing protein catabolism (e.g. starvation), therefore in the increased supply of amino acids. The urea cycle belongs among proton-producing reactions, its activity is reduced at lower pH – acidosis.
Glucose-alanine cycle
Alanine participates in the transmission of blood ammonia and also serves through pyruvate as a significant source of carbons in gluconeogenesis – see Subchapter 2/9. Glucose-alanine cycle is pathway extending between the liver and muscle cells. Pyruvate produced in muscle cells undergoes the transamination to give alanine. It is released into the blood and transferred to the liver where it is converted back to pyruvate by transamination that can be involved in the process of gluconeogenesis. The resulting glucose is released into the blood and enters muscle cells and the cycle continues. Transferred amino group (ammonia) is directed to the urea cycle.
_
Degradation of amino acid carbon skeletons
Proteins in the human body contain 20 (21 if we include selenocysteine) proteinogenic amino acids. Twenty (twenty-one) different multienzyme sequences exist for catabolism of amino acid carbon skeletons. In this text we restrict ourselves only to the basic general mechanisms of amino acid carbon skeleton degradation and a few selected examples.
Catabolism of amino acid carbon skeletons results in the formation of seven products: pyruvate, acetyl-CoA, acetoacetyl-CoA, α-ketoglutarate, suc-CoA, fumarate and oxaloacetate. They have a different fate in the energy metabolism. The strategy of the cell is to convert amino acid carbon skeletons to compounds useful in gluconeogenesis or a molecule of lipids (fatty acids and ketone bodies). Amino acids are divided into glucogenic and ketogenic amino acids according to the fate of their degradation products. Amino acids leucine and lysine (starting with the letter L) belong among ketogenic amino acids which lead to the formation of acetyl-CoA and acetoacetyl-CoA. Glucogenic amino acids include those that lead to the formation of the remaining five products – pyruvate, α-ketoglutarate, suc-CoA, fumarate or oxaloacetate – serine, threonine, cysteine, methionine, aspartate, glutamate, asparagine, glutamine, glycine, alanine, valine, proline, histidine and arginine. But some amino acids have two degradation products – one of them being glucogenic and second one ketogenic. These amino acids are called keto- and glucogenic amino acids – they include isoleucine, phenylalanine, tyrosine and tryptophan.
The following overview shows degradation products of particular amino acids:
1) Acetyl-CoA and acetoacetyl-CoA – Lys and Leu are purely ketogenic amino acids, some other amino acids (Phe, Tyr, Trp, Ile) provide glucogenic and ketogenic degradation products
2) α-ketoglutarate – five-carbon amino acids – Glu, Gln, Pro, Arg a His
3) Suc-CoA – nonpolar amino acids – Met, Ile a Val
4) Fumarate – Phe, Tyr
5) Oxaloacetate – four-carbon amino acids – Asp a Asn
6) Pyruvate – Cys, Ala, Ser, Gly, Thr, Trp
Degradation of branched amino acids – Val, Leu and Ile
These amino acids are not degraded in the liver cells, but mainly in extrahepatic tissues – especially high activity in muscle cells. They contain specific transaminase producing the appropriate α-keto acids – the so-called keto analogs of branched amino acids. This transaminase is not present in liver cells. Keto analogs are converted to acyl-CoA derivatives by a dehydrogenation complex which catalyzes the oxidative decarboxylation and dehydrogenation.
The genetic defect of dehydrogenation complex causes a maple syrup urine disease. This relatively rare disease can lead to the accumulation of the appropriate α-keto acids in tissues and body fluids (urine smells like caramel). The defect causes abnormal brain development, mental retardation and can result in death of the individual.
_
Formation of non-essential amino acids in the human body
The human body is not able to synthesize essential amino acids – Phe, Trp, Val, Leu, Ile, Met, Thr and Lys. Two amino acids are essential in the growth of the organism (the period of their increased consumption), the rate of their synthesis is not sufficient to cover the claims of the body – the so-called conditionally essential amino acids – Arg, His. Other amino acids belong to non-essential amino acids. Here is the overview of amino acid precursors:
1) Oxaloacetate → Asp, Asn
2) α-ketoglutarate → Glu, Gln, Pro, (Arg and His)
3) Pyruvate → Ala
4) 3-phosphoglycerate → Ser, Cys and Gly
5) Phe → Tyr
Phenylketonuria (PKU)
Phenylketonuria (PKU) is an autosomal recessive metabolic disorder (incidence 8-10 cases / 100 000 individuals) conditioned by the absence or reduced phenylalanine hydroxylase activity. Physiologically this enzyme catalyzes hydroxylation of Phe to Tyr. The defective enzyme leads to the phenylalanine accumulation and an alternative degradation of Phe – phenylpyruvate (transamination), phenyllactate, phenylacetate and phenylethylamine are formed. Such substances accumulate in tissues and body fluids and produce typical odor of urine (mousy smell). Some of them cause severe brain damage. Phenylketonuria was the first discovered human genetic defect in amino acid metabolism and is currently one of the diseases for which a screening is carried out in all newborns. If it is detected even at this age, we can prevent brain damage with a special diet low in Phe.
_
Important derivatives of individual amino acids
Decarboxylation – formation of biogenic amines
Some amino acids undergo decarboxylation (removal of carboxyl group). The result is the formation of biogenic amines (monoamines) that exhibit a broad spectrum of functions in the human body. Here is the basic outline:
1) Tyr → catecholamines (DOPA →dopamine → noradrenaline (norepinephrine) → adrenaline (epinephrine)
2) Trp → serotonin (5-hydroxytryptamine)
3) Glu → γ-aminobutyrate (GABA)
4) His → histamine
5) Ser → ethanolamine → choline → acetylcholine
6) Cys → cysteamine
7) Asp → β-alanine
Nitric oxide (NO)
Nitric oxide is a vasodilator substance produced by endothelial cells. It is formed from L-arginine in the reaction catalyzed by nitric oxide synthase (NO-synthase):
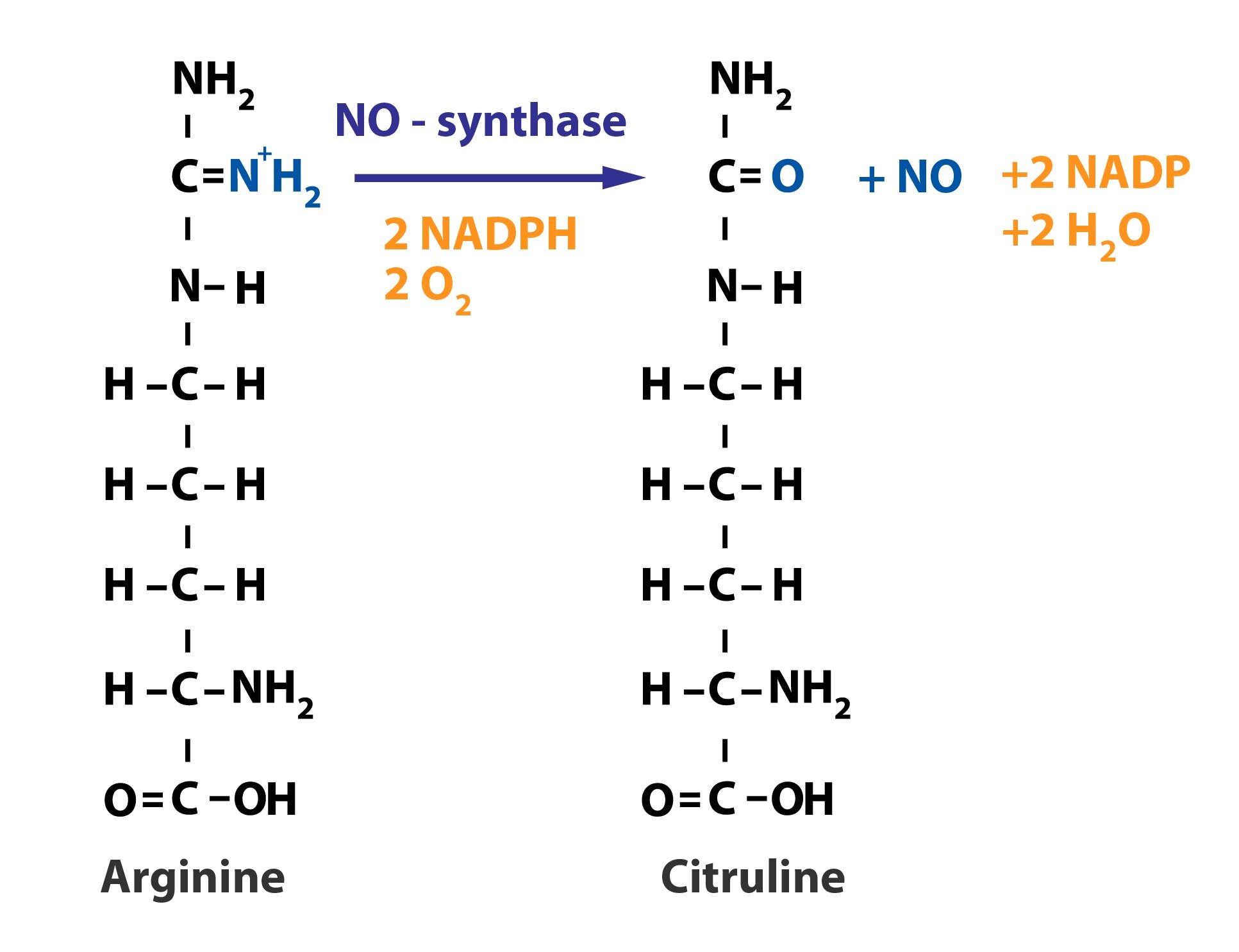
NO-synthase is present not only in endothelial cells but also in some cells of the immune system (one of the cytotoxic agents), and in certain neurons. The mechanism and significance of NO-synthase action are described in Subchapter 10/2 and Subchapter 5/3.
Other important derivatives of amino acids
Trp → melatonin
Phe and Tyr → thyroid hormones, melanin
Gly → heme, purines, creatine, conjugation with bile acids
Arg and Ornithine → creatine, polyamines (spermidine, spermine)
Gly, Glu, Asp → purines and pyrimidines
Cys → taurine
_
Subchapter Authors: Josef Fontana and Petra Lavríková
![]()